What evidence is there to support that there is a genetic basis for language?
Introduction
Specific language impairment (SLI) is a developmental language disorder characterized by impaired oral linguistic communication skills (Leonard et al., 1999; Catts et al., 2005). The disorder is typically diagnosed in the preschool years, when children unremarkably brainstorm speaking in more complex and consummate sentences. These children take normal non-verbal IQ in spite of their bug with semantics, syntax, and discourse (Paul, 2007). Authentication grammatical errors include the omission of articles (such as "the"), pronoun mistakes (e.yard., "him" in place of "he"), grammatical inflection (e.g., "go" instead of "goes"), and tense errors (e.1000., switching present for past tense).
Children with language impairments rarely have a single gene mutation and information technology is agreed that fifty-fifty individuals with the same disorder are unlikely to have the exact same prepare of genetic markers (Bishop, 2002). The lack of a consistent causal cistron has led some to speculate that circuitous developmental disorders such as dyslexia, attention deficits, and SLI are instead due to any one of several combinations of genetic markers. Recently, evidence has suggested that some genetic variants may create a susceptibility to developmental disorders, and these susceptibility variants may exist common in many individuals, even if the rest of their genetic variants differ (Donlon, 1988; Wang et al., 2009; Burnside et al., 2011; Centanni et al., 2015; Hashemi et al., 2015).
The effect of copy number variants (CNVs) on chromosome fifteen (q11.2) has been a field of study of argue in the field, both anecdotally and in scholarly articles. Microdeletions and microduplications in this region accept been associated with a variety of disorders, including autism, schizophrenia, Prader–Willi, and Angelman's syndromes (Kirov et al., 2009; Hogart et al., 2010; Mefford et al., 2010; Dimitropoulos et al., 2013; Hashemi et al., 2015). However, duplications at this location are commonly seen in typically developing individuals (Mefford et al., 2010), which raises questions well-nigh whether variants at this location play a role in the disorders mentioned above. The consistent association betwixt this region and a variety of developmental disorders suggests that variants in this region do contribute to the disordered land, even if they are non causal on their own. Though re-create number variations in this region have been suggested equally a susceptibility variant in many disorders, it is currently unknown if these variants are susceptibility factors for disorders such as SLI.
In the current report, we discuss the behavioral and genetic profiles of eight children with SLI who took part in a larger study on the biological footing of language impairment. Due to the current controversy regarding the definition of SLI and its diagnostic criterion (Reilly et al., 2014), we used strict assessment score cutoffs that are in line with other studies on the genetics of SLI (Rice et al., 2009). 4 of these children all had gains in this region of chromosome 15 also as additional CNVs in multiple other regions previously linked to language impairments.
Materials and Methods
Participants
In the current study, we discuss iv children who were part of a cohort of viii children with SLI, ranging in historic period from four;5 to 17;2 (years;months), that participated in a larger study on the biological pathways of speech and language disorders. All procedures were approved by the Institutional Review Board of the University of Nebraska Medical Centre and all participants were consented prior to participation. Participants completed a series of usually administered, age-appropriate speech, linguistic communication, reading, and cognitive assessments including the Goldman Fristoe Examination of Joint-Second Edition (GFTA-2; Goldman and Fristoe, 2000), the Clinical Evaluation of Linguistic communication Fundamentals-Fourth Edition (CELF-4; Semel et al., 2003), Reynolds Intellectual Assessment Scales (RIAS; Reynolds and Kamphaus, 2005), and the Woodcock Reading Mastery Test-Revised (WRMT-R; Woodcock, 1998). All participants were required to have normal cognition based on a standard score college of 75 or higher on the RIAS.
Children were assigned to the SLI group based on GFTA-2 percentile scores of xvi or college and a CELF-4 standard score below 85. Inclusionary criteria are presented in Tabular array 1.

Tabular array 1. Inclusionary benchmark for categorization in the specific language impairment (SLI) group.
Deoxyribonucleic acid Collection and Isolation
Buccal cell samples were collected from all eight participants with SLI using the Isohelix DNA swab packs (Cell Projects, Ltd., Kent, UK), and Deoxyribonucleic acid was extracted per manufacturer'southward recommendations using the QIACube (Qiagen, Valencia, CA, U.s.). Dna quantity and quality were determined using the NanoDrop ND-1000® spectrophotometer (NanoDrop Technologies, Wilmington, DE, United states of america) and agarose gel electrophoresis, respectively.
CNV Detection
High-resolution genome-wide analysis was performed on genomic Deoxyribonucleic acid using the CytoScanHDTM array (Affymetrix, Santa Clara, CA, USA) according to manufacturer'south instruction. This array contains more 2.vi million markers for high-resolution whole-genome copy number analysis and 750,000 genotype-able single nucleotide polymorphisms (SNPs) for reliable detection of copy neutral loss of heterozygosity (CN-LOH). Data were visualized and analyzed with the Chromosome Analysis Suite (ChAS) software (Affymetrix) using the following filter parameters: (ane) ≥25 markers and ≥5 kilobases (kb) for CNVs and (2) ≥5 megabases (Mb) for CN-LOH. All basepairs are mapped to Build 37/hg19. Parental Deoxyribonucleic acid samples were non available for these children, so it was not possible to determine whether these were de novo variants.
Statistical Assay
We used Pearson's correlation to evaluate the relationship between gain size in 15q11.ii and phenotype characteristics (p < 0.05).
Results
Behavioral Profile
All eight children were administered a number of speech, linguistic communication, and cognitive assessments to ensure a diagnosis of SLI in the absenteeism of any comorbid weather condition (Tabular array i). All children were classified equally having SLI since they scored below 85 on the language measure in the presence of normal non-verbal IQ and no articulation impairments (Table ii). Because children five through viii did not evidence any variants at 15q11.two, their data were excluded from further consideration in this paper. Child ane was x;3 (years;months) and female, child two was 9;three and female, child 3 was 10;ane and female, and child four was eleven;3 and male person. None of the children scored within the dumb range on the word reading measure (<85), only they did brandish a wide range of typical discussion reading abilities, from the 30th percentile (child ane, 30; child ii, 32; kid 3, 45) upward to the 73rd percentile (child 4).

Tabular array 2. Assessment standard scores (percentiles in parenthesis where applicative) for four children with copy number variants (CNVs) at 15q11.2.
Genetic Profile
Of the eight children with SLI that were genotyped, four of these children had big gains in an overlapping region at 15q11.2. Child 1's gain was 54.34 kilo-bases in length (25283093–25337431), child two had a gain of 41.70 kb (25295728–25337431), child 3 had a gain of 32.94 kb (25291742–25324677), and child iv had a gain of 14.81 kb (25306864–25321675). These gains are large and embrace a variety of genes. Because of the size of these gains, exons and introns for a diverseness of genes were affected. About hits amid these iv children included the genes SNORD109A, SNORD109B, and SNORD116-(1-23). These genes are usually associated with Prader–Willi syndrome, with evidence suggesting that several genes, including SNORD116, are a primal pathogenic component (Rabinovitz et al., 2012; Anderlid et al., 2014).
We too annotated copy number at other known linguistic communication, and more broadly, neurodevelopmental loci. Overall, the range of cess scores and the multifariousness of proceeds sizes in the sample suggest that other genetic factors may be contributing to the observed phenotypes (Table iii). Child i had 2 additional significant gains or losses (time to come collectively chosen 'hits'). The showtime was a gain at 13q21.1, which has been seen in individuals with autism and language harm (Bartlett et al., 2004). The second was a loss at 12p13.33, which has been previously associated with childhood apraxia of speech (CAS) and attention difficulties (Thevenon et al., 2013). Child 2 had 2 hits of clinical significance in addition to 15q11.two. The commencement was a loss at 10q21.1, which has been associated with intellectual inability, lack of expressive speech, and attending deficit and hyperactivity disorder (ADHD; Neale et al., 2010; Freunscht et al., 2013). The 2d was a loss at 16p11.ii, which has been previously associated with autism (Kumar et al., 2008; Weiss et al., 2008; Laffin et al., 2012). This gene is oft characterized as pathogenic and likely contributed to the phenotype of this child.

Tabular array 3. Additional CNVs for each of four children with hits at 15q11.2.
Child iii had ii CNVs in addition to the deletion at 15q11.2, both of which may exist pathogenic. The first was a loss at 9p24.3 involving the gene DOCK8 and has been previously linked with intellectual inability (de Vries et al., 2005; Griggs et al., 2008). In fact, this child did have the lowest non-verbal IQ (standard score of 98) of the four children described hither (Table 2). The second was a gain at 22q13.33. A duplication of this region is linked with developmental delay and the region also contains the gene SHANK3, which has been associated with autism (Durand et al., 2007; Moessner et al., 2007). Finally, kid 4 had i CNV in addition to the deletion at 15q11.2: a gain at 7q11.23, which has been associated with language delay and the Williams–Beuren locus (Jurado et al., 1998; Somerville et al., 2005; Berg et al., 2007). The result that these children all exhibited other genetic variants previously associated with speech and linguistic communication impairments support the idea that a variant at 15q11.two is a susceptibility locus and not necessarily one that is deleterious past itself.
The genetic profiles have an interesting phenotypic context for evaluating the office of 15q11.2 relative to reading and language. We consider the clustering of percentiles for children i–3 and the relative outlier of child iv. Kid 4 had the smallest gain (eighteen.13 kb less than the next largest proceeds, in kid three) and likewise had the highest scores on the linguistic communication and word reading measures. Although this child is the oldest in our sample, information technology is unlikely that historic period was a factor considering that the reading scores were normed for age. Although there are merely four data points, there is a significant linear association between the size of the gain observed and the scores on the discussion reading measure (r = -0.96, p = 0.04; Figure 1). In spite of the age correction, Kid 4's data point could be a possible outlier. Hereafter studies with a larger group of children are required to validate this potential association between proceeds size and word reading scores.
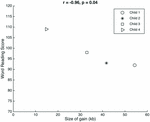
FIGURE 1. Relation betwixt gain size and word reading. There was a significant correlation between the size of the proceeds observed at 15q11.2 in each of the children and their respective scores on the word reading measure.
Prevalence of Observed Hits in the General Population
Search of the Database of Genomic Variants (DGV; http://dgv.tcag.ca/) yielded ane unselected sample with variation at 15q11.2. A single deletion was found (1 out of N = 873 subjects) in the Ontario Population Genomics Platform (OPGP) controls (Costain et al., 2013). This study used the same technology as our study (Affymetrix-CytoScanHD). To evaluate the population prevalence of the secondary hits observed in our four children, we likewise searched the DGV and the OPGP for each of the variants reported here. Population frequencies in these ii populations are shown in Table 3.
Discussion
In the current study, nosotros study the behavioral phenotypes of four children with SLI who too had large gains in the q11.2 region of chromosome 15. These children all had poor oral language abilities compared to typical peers. In spite of normal not-verbal intelligence and normal spoken communication joint, these children had a wide range of abilities in word reading. Three of the iv children also had boosted genetic variants located in areas previously associated with speech and language impairments. These results support the theory that variants at 15q11.ii may create an increased predisposition to displaying a language disorder.
Strengths and Caveats of the Current Study
A strength of the electric current study is the strict benchmark used to identify children with SLI. Considering this disorder oft co-occurs with dyslexia (Leonard et al., 2002; McCarthy et al., 2012), it has been difficult to determine which genes are related to SLI specifically and which are related to dyslexia. Though our sample size was small (eight children with SLI), the event that four of the eight had a duplication at 15q11.2 supports previous work linking developmental disorders with microdeletions or microduplications in this region (de Kovel et al., 2010; Hashemi et al., 2015). Since all the children in our study were confirmed as having SLI, we were unable to provide back up for previous reports that this CNV tin can occur in typically developing individuals. Time to come studies should investigate this mark in a larger population of typically developing children as well as those with SLI in the absence of comorbid conditions.
The Multiple Hits Model of Developmental Disorders
To date, no single genetic marker reliably predicts the occurrence of SLI. Information technology is likely that SLI, and perhaps other communication disorders, are caused by a constellation of genes (Rice et al., 2009). An existing hypothesis states that region 15q11.two is a susceptibility variant. If so, a hit in this region could increase the likelihood that an individual will showroom a developmental disorder phenotype when additional risk variants are also present. Microdeletions in this region are commonly associated with developmental disorders similar Prader–Willi Syndrome (Dimitropoulos et al., 2013) and Angelman Syndrome (Donlon, 1988), as well as epilepsy (Mefford et al., 2010) and autism (de Kovel et al., 2010). For example, a microdeletion in 15q11.2 was observed in 1% of individuals with idiopathic generalized epilepsies (12 of 1234; de Kovel et al., 2010). These deletions are often seen in unaffected family unit members in addition to affected offspring.
Though the variants seen in the electric current study were microduplications rather than microdeletions, recent bear witness suggests that this blazon of variant may as well betoken susceptibility to developmental disorders, including autism (Hogart et al., 2010; Kitsiou-Tzeli et al., 2010; van der Zwaag et al., 2010) and speech delay (Burnside et al., 2011). The consistent ascertainment that microduplications in 15q11.two are associated with SLI in our sample, together with previous evidence that microdeletions in this area are related to other developmental disorders, suggests that this region is sensitive to mutations of various forms. It is interesting to note that the iv children with gains at 15q11.2 did not testify any variants in regions previously associated with SLI, including 16q (SLI1), 19q (SLI2), and 13q (SLI3) (Bartlett et al., 2004; Consortium, 2004; Monaco, 2007; Newbury et al., 2011). Specifically, a locus at 16q known as SLI1, has not only been linked with SLI in a large sample, just is likewise associated with basic reading, spelling, and reading comprehension measures (Consortium, 2004). The observation that none of our participants exhibited variants in these notable regions is probable due to a combination of study design and sample size. Developmental linguistic communication and communication disorders are notorious for having a complicated genetic picture, without a unmarried causal cistron (Bishop, 2002). Information technology is possible that the variant at 15q represents another path to SLI in the absence of variants at the previously associated areas.
Our event provides additional support to 15q11.2 as a susceptibility locus, though larger studies of persons with linguistic communication and related cognitive phenotypes are needed to establish the prevalence of this variant in the general population compared with a diverseness of developmental disorders.
Conflict of Interest Statement
The authors declare that the inquiry was conducted in the absence of any commercial or financial relationships that could be construed equally a potential conflict of interest.
Acknowledgments
This research was supported past the University of Nebraska Health Research Consortium (Co-PIs Hogan and Green) and the Barkley Memorial Trust. The authors wish to thank Jennifer Sanmann and Warren Sanger, for their expertise in genetic data analysis and their invaluable comments on previous versions of this manuscript. Thank you also to the post-obit individuals: Kimber Green, Sara Benham, Dyann Rupp, Tacy Corson, Phoebe Chung, Natalie Vanderveen, and Kristin Schneller for their assistance with data collection and Diane Pickering and Danielle Bishay for specimen processing and genetic data analysis.
References
Anderlid, B., Lundin, J., Malmgren, H., Lehtihetm, M., and Nordgren, A. (2014). Pocket-sized mosaic deletion encompassing the snoRNAs and SNURF–SNRPN results in an atypical Prader–Willi syndrome phenotype. Am. J. Med. Genet. 164A, 425–431. doi: 10.1002/ajmg.a.36307
PubMed Abstract | CrossRef Full Text | Google Scholar
Bartlett, C., Flax, J., Logue, M., Smith, B., Vieland, V. J., Tallal, P., et al. (2004). Test of potential overlap in autism and language loci on chromosomes 2, vii, and 13 in two contained samples ascertained for specific linguistic communication harm. Hum. Hered. 57, 10–twenty. doi: 10.1159/000077385
PubMed Abstract | CrossRef Full Text | Google Scholar
Berg, J., Brunetti-Pierri, N., Peters, S. Kang, Due south. H., Fong, C. T., Salamone, J., et al. (2007). Oral communication delay and autism spectrum behaviors are often associated with duplication of the 7q11. 23 Williams-Beuren syndrome region. Genet. Med. ix, 427–441. doi: 10.1097/GIM.0b013e3180986192
PubMed Abstract | CrossRef Total Text | Google Scholar
Bishop, D. (2002). The role of genes in the etiology of specific language impairment. J. Commun. Disord. 35, 311–328. doi: 10.1016/S0021-9924(02)00087-4
CrossRef Full Text | Google Scholar
Burnside, R., Pasion, R., Mikhail, F., Carroll, A. J., Robin, Due north. H., Youngs, Due east. 50., et al. (2011). Microduplication of proximal 15q11. 2 betwixt BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language filibuster. Hum. Genet. 130, 517–528. doi: 10.1007/s00439-011-0970-4
PubMed Abstract | CrossRef Full Text | Google Scholar
Catts, H. W., Adlof, S. M., Hogan, T. P., and Weismer, Southward. Eastward. (2005). Are Specific Language Impairment and Dyslexia Singled-out Disorders? J. Speech communication Lang. Hear. Res. 48, 1378–1396. doi: 10.1044/1092-4388(2005/096)
CrossRef Total Text | Google Scholar
Centanni, T., Sanmann, J., Green, J. R., Iuzzini-Seigel, J., Bartlett, C., Sanger, West. G., et al. (2015). The function of candidate–gene CNTNAP2 in childhood apraxia of speech and specific language impairment. Am. J. Med. Genet. B Neuropsychiatr. Genet. doi: 10.1002/ajmg.b.32325 [Epub alee of print].
PubMed Abstract | CrossRef Full Text | Google Scholar
Consortium, South. (2004). Highly significant linkage to the SLI1 locus in an expanded sample of individuals affected past specific language damage. Am. J. Hum. Genet. 74, 1225–1238. doi: 10.1086/421529
PubMed Abstruse | CrossRef Full Text | Google Scholar
Costain, One thousand., Lionel, A. C., Merico, D., Forsythe, P., Russell, K., Lowther, C., et al. (2013). Pathogenic rare copy number variants in community-based schizophrenia suggest a potential role for clinical microarrays. Hum. Mol. Genet. 22, 4485–4501. doi: 10.1093/hmg/ddt297
PubMed Abstruse | CrossRef Full Text | Google Scholar
de Kovel, C., Trucks, H., Helbig, I., Mefford, H., Baker, C., Leu, C., et al. (2010). Recurrent microdeletions at 15q11. 2 and 16p13. xi predispose to idiopathic generalized epilepsies. Brain 133(Pt 1), 23–32. doi: 10.1093/brain/awp262
PubMed Abstract | CrossRef Full Text | Google Scholar
de Vries, B. B. A., Pfundt, R., Leisink, One thousand., Koolen, D. A., Vissers, 50. E. L. M., Janssen, I. Grand., et al. (2005). Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 77, 606–616. doi: 10.1086/491719
PubMed Abstract | CrossRef Full Text | Google Scholar
Dimitropoulos, A., Ferranti, A., and Lemler, Grand. (2013). Expressive and receptive language in Prader-Willi syndrome: report on genetic subtype differences. J. Commun. Disord. 46, 193–201. doi: 10.1016/j.jcomdis.2012.12.001
PubMed Abstract | CrossRef Total Text | Google Scholar
Donlon, T. (1988). Like molecular deletions on chromosome 15q11. 2 are encountered in both the Prader-Willi and Angelman syndromes. Hum. Genet. 80, 322–328. doi: 10.1007/BF00273644
PubMed Abstract | CrossRef Full Text | Google Scholar
Durand, C., Betancur, C., Boeckers, T., Bockmann, J., Celibate, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
PubMed Abstract | CrossRef Full Text | Google Scholar
Freunscht, I., Popp, B., Bare, R., Endele, Southward., Moog, U., Petri, H., et al. (2013). Behavioral phenotype in v individuals with de novo mutations within the GRIN2B gene. Behav. Brain Funct. 9, twenty. doi: 10.1186/1744-9081-9-20
PubMed Abstract | CrossRef Full Text | Google Scholar
Goldman, R., and Fristoe, Thousand. (2000). Goldman-Fristoe Examination of Articulation-ii (GFTA-2). Circumvolve Pines, MN: American Guidance Service.
Google Scholar
Griggs, B. L., Ladd, Southward., Saul, R. A., DuPont, B. R., and Srivastava, A. K. (2008). Dedicator of cytokinesis 8 is disrupted in ii patients with mental retardation and developmental disabilities. Genomics 91, 195–202. doi: 10.1016/j.ygeno.2007.10.011
PubMed Abstruse | CrossRef Full Text | Google Scholar
Hashemi, B., Bassett, A., Chitayat, D., Chong, Grand., Feldman, M., Flanagan, J., et al. (2015). Deletion of 15q11.two(BPone-BP2) region: further evidence for lack of phenotypic specificity in a pediatric population. Am. J. Med. Genet. A doi: ten.1002/ajmg.a.37134 [Epub ahead of print].
PubMed Abstract | CrossRef Full Text | Google Scholar
Hogart, A., Wu, D., LaSalle, J. M., and Schanen, North. C. (2010). The comorbidity of autism with the genomic disorders of chromosome 15q11.2–q13. Neurobiol. Dis. 38, 181–191. doi: 10.1016/j.nbd.2008.08.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Jurado, 50., Wang, Y. Thou., Peoples, R., Coloma, A., Cruces, J., and Francke, U. (1998). A duplicated gene in the breakpoint regions of the 7q11. 23 Williams-Beuren syndrome deletion encodes the initiator binding poly peptide TFII-I and BAP-135, a. Hum. Mol. Genet. seven, 325–334. doi: 10.1093/hmg/7.3.325
PubMed Abstract | CrossRef Full Text | Google Scholar
Kirov, 1000., Grozeva, D., Norton, North., Ivanov, D., Mantripragada, Chiliad. K., Holmans, P., et al. (2009). Support for the involvement of large re-create number variants in the pathogenesis of schizophrenia. Hum. Mol. Genet. 18, 1497–1503. doi: 10.1093/hmg/ddp043
PubMed Abstruse | CrossRef Full Text | Google Scholar
Kitsiou-Tzeli, S., Tzetis, M., Sofocleous, C., Vrettou, C., Xaidara, A., Giannikou, K., et al. (2010). De novo interstitial duplication of the 15q11.2-qxiv PWS/AS region of maternal origin: clinical description, array CGH assay, and review of the literature. Am. J. Med. Genet. A 152A, 1925–1932. doi: 10.1002/ajmg.a.33447
PubMed Abstruse | CrossRef Full Text | Google Scholar
Kumar, R., KaraMohamed, Due south., Sudi, J., Conrad, D. F., Brune, C., Badner, J. A., et al. (2008). Recurrent 16p11. 2 microdeletions in autism. Hum. Mol. Genet. 17, 628–638. doi: x.1093/hmg/ddm376
PubMed Abstruse | CrossRef Full Text | Google Scholar
Laffin, J. J. S., Raca, Thousand., Jackson, C. A., Strand, E. A., Jakielski, K. J., and Shriberg, L. D. (2012). Novel candidate genes and regions for babyhood apraxia of speech identified by array comparative genomic hybridization. Genet. Med. 14, 928–936. doi: 10.1038/gim.2012.72
PubMed Abstract | CrossRef Full Text | Google Scholar
Leonard, C., Lombardino, L., Walsh, K., Eckert, M. A., Mockler, J. L., Rowe, L. A., et al. (2002). Anatomical risk factors that distinguish dyslexia from SLI predict reading skill in normal children. J. Commun. Disord. 35, 501–531. doi: 10.1016/S0021-9924(02)00120-Ten
PubMed Abstract | CrossRef Full Text | Google Scholar
Leonard, L., Miller, C., and Gerber, E. (1999). Grammatical morphology and the lexicon in children with specific linguistic communication harm. J. Speech Lang. Hear. Res. 42, 678–689. doi: 10.1044/jslhr.4203.678
CrossRef Full Text | Google Scholar
McCarthy, J. H., Hogan, T. P., and Catts, H. Westward. (2012). Is weak oral language associated with poor spelling in school-age children with specific language damage, dyslexia or both? Clin. Linguist. Phonet. 26, 791–805. doi: x.3109/02699206.2012.702185
PubMed Abstruse | CrossRef Full Text | Google Scholar
Mefford, H. C., Muhle, H., Ostertag, P., von Spiczak, Due south., Buysse, Yard., Baker, C., et al. (2010). Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 6:e1000962. doi: 10.1371/journal.pgen.1000962
PubMed Abstract | CrossRef Full Text | Google Scholar
Moessner, R., Marshall, C., Sutcliffe, J., Skaug, J., Pinto, D., Vincent, J., et al. (2007). Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Med. Genet. 81, 1289–1297. doi: 10.1086/522590
PubMed Abstract | CrossRef Total Text | Google Scholar
Neale, B. M., Medland, S., Ripke, South., Anney, R. J. L., Asherson, P., Buitelaar, J., et al. (2010). Case-control genome-wide association study of attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry 49, 906–920. doi: 10.1016/j.jaac.2010.06.007
PubMed Abstract | CrossRef Full Text | Google Scholar
Newbury, D. F., Paracchini, S., Scerri, T. South., Winchester, L., Addis, 50., Richardson, A. J., et al. (2011). Investigation of dyslexia and SLI risk variants in reading-and language-dumb subjects. Behav. Genet. 41, 90–104. doi: 10.1007/s10519-010-9424-3
PubMed Abstract | CrossRef Full Text | Google Scholar
Rabinovitz, S., Kaufman, Y., Ludwig, Yard., Razin, A., and Shemer, R. (2012). Mechanisms of activation of the paternally expressed genes by the Prader-Willi imprinting center in the Prader-Willi/Angelman syndromes domains. Proc. Natl. Acad. Sci. U.S.A. 109, 7403–7408. doi: 10.1073/pnas.1116661109
PubMed Abstract | CrossRef Full Text | Google Scholar
Reilly, Southward., Tomblin, B., Police, J., McKean, C., Mensah, F. K., Morgan, A., et al. (2014). Specific linguistic communication impairment: a user-friendly label for whom? Int. J. Lang. Commun. Disord. 49, 416–451. doi: 10.1111/1460-6984.12102
PubMed Abstruse | CrossRef Full Text | Google Scholar
Reynolds, C. R., and Kamphaus, R. W. (2005). "Introduction to the Reynolds intellectual cess scales and the Reynolds intellectual screening exam," in Gimmicky Intellectual Assessment: Theories, Tests, and Problems, second Edn, eds D. P. Flanagan and P. Fifty. Harrison (New York, NY: Guilford Press), 461–483.
Google Scholar
Rice, Thousand. L., Smith, S. D., and Gayán, J. (2009). Convergent genetic linkage and associations to language, speech and reading measures in families of probands with specific language harm. J. Neurodev. Disord. 1, 264–282. doi: 10.1007/s11689-009-9031-10
PubMed Abstract | CrossRef Full Text | Google Scholar
Semel, Eastward., Wiig, Due east., and Secord, West. (2003). Clinical Evaluation of Language Fundamentals-IV. Marickville: Harcourt Assessment.
Google Scholar
Somerville, M. J., Mervis, C. B., Immature, East. J., Seo, E.-J., del Campo, Yard., Bamforth, S., et al. (2005). Severe expressive-linguistic communication delay related to duplication of the Williams-Beuren locus. N. Engl. J. Med. 353, 1694–1701. doi: x.1056/NEJMoa051962
PubMed Abstract | CrossRef Total Text | Google Scholar
Thevenon, J., Callier, P., Andrieux, J., Delobel, B., David, A., Sukno, S., et al. (2013). 12p13.33 microdeletion including ELKS/ERC1, a new locus associated with childhood apraxia of speech. Eur. J. Hum. Genet. 21, 82–88. doi: 10.1038/ejhg.2012.116
PubMed Abstract | CrossRef Full Text | Google Scholar
van der Zwaag, B., Staal, W. G., Hochstenbach, R., Poot, M., Spierenburg, H. A., de Jonge, Chiliad. V., et al. (2010). A co-segregating microduplication of chromosome 15q11.ii pinpoints two risk genes for autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiat. Genet. 153B, 960–966. doi: 10.1002/ajmg.b.31055
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, One thousand., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., et al. (2009). Common genetic variants on 5p14. ane associate with autism spectrum disorders. Nature. 459, 528–533. doi: 10.1038/nature07999
PubMed Abstract | CrossRef Full Text | Google Scholar
Weiss, L., Shen, Y., Korn, J., Arking, D. E., Miller, D. T., Fossdal, R., et al. (2008). Association betwixt microdeletion and microduplication at 16p11. 2 and autism. Engl. J. Med. Bachelor at: http://world wide web.nejm.org/doi/total/x.1056/NEJMoa075974
Google Scholar
Woodcock, R. Due west. (1998). Woodcock Reading Mastery Tests, Revised. Circle Pines, MN: American Guidance Service.
Google Scholar
Source: https://www.frontiersin.org/articles/153884
{kind=link}
Post a Comment for "What evidence is there to support that there is a genetic basis for language?"